

背景介绍
低温严重损害了锂离子电池的性能,因此锂离子电池需要具有宽流动性范围、离子扩散速度快和脱溶剂化能较低的电解质。而这里面的关键在于在Li+和溶剂分子之间建立温和的内部相互作用,这在商业碳酸亚乙酯基电解质中很难实现。
正文部分
成果简介
近日,湖南大学刘继磊教授,构建了低介电常数(ε)溶剂主导配位的溶剂化结构,并通过羰基氧的电负性调节碳酸亚乙酯的配位强度。改性电解质在−90 °C时表现出高离子电导率(1.46 mS·cm−1) ,并在−110 °C时保持液态。因此,4.5 V石墨基软包电池在−10 °C的200次循环中达到约98%的容量 保持率,没有锂枝晶。这些电池在−70 °C时也能保持约60%的室温放电容量,即使在约−100 °C的温度下也能奇迹般地保持放电功能。该研究以题目为“Breaking solvation dominance of ethylene carbonate via molecular charge engineering enables lower temperature battery”的论文发表在国际顶级期刊《Nature Communications》上。

图文导读
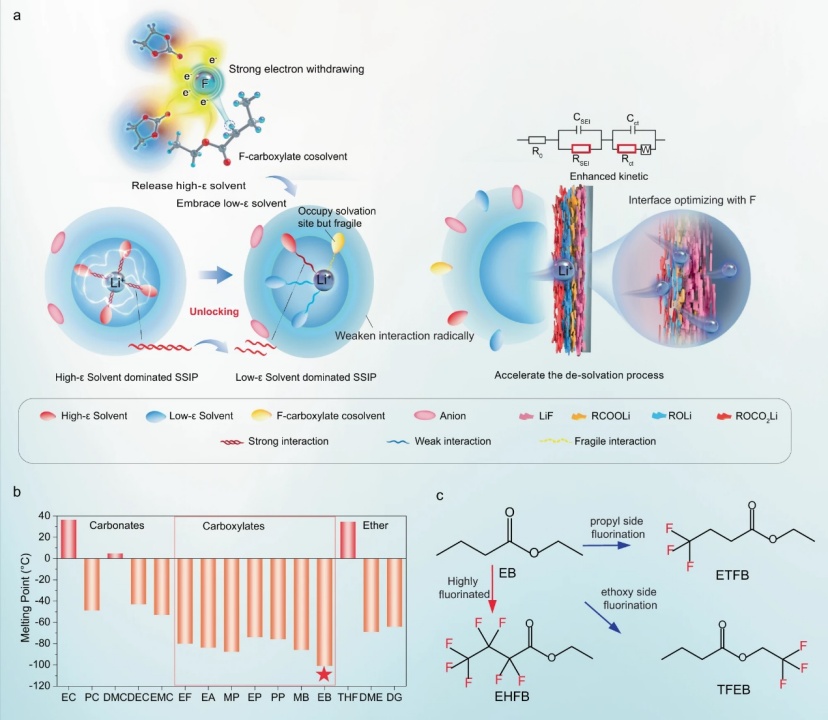
【图1】a低温电解质设计原则。b常见碳酸盐和羧酸盐溶剂的熔点。c所选EB及其氟化类似物共溶剂的化学结构。
在LIB电解质中,Li+通常通过电负性羰基氧与碳酸盐电解质中的极性溶剂分子配位。理论上,在不牺牲极性溶剂的高ε性质的情况下削弱这种配位的一种方法是降低高ε溶剂中羰基氧的电负性,而不是用低ε溶剂取代它。这可能通过引入强吸电子元素来实现,例如氟。氟化预计会削弱高ε溶剂(即环状碳酸酯)和Li+之间的配位相互作用(图1a),导致(i)释放更多的高ε溶剂,这将促进低ε溶剂(如线性碳酸酯)与Li+的配位,占据其配位位点,从而将溶剂分离的离子对(SSIP)从高ε溶剂为主转化为低ε溶剂为主,以及(ii)打破剩余配位的高ε溶剂的相互作用(图1a)。独特的溶剂化结构具有几个优点:(i)通过降低高ε溶剂的配位数和强度以及促进低ε溶剂主导的溶剂化结构的形成,显著而彻底地削弱了Li+和溶剂之间的整体配位相互作用(图1a)。此外,氟化羧酸盐共溶剂也将通过羰基参与溶剂化,即使其与Li+的相互作用减弱(图1a)。所有这些变化一起促进了所需溶剂化结构的形成,其中Li+和所有溶剂之间的相互作用弱得多,这有望拓宽液体温度范围和进一步促进Li+脱溶剂化。(ii)由于较低的LUMO能量,氟化共溶剂将有利于富F SEI的形成。此外,氟化会降低电解质的HOMO能量,从而拓宽其在高压电池中的电化学窗口。这种方法提供了一种更有效的方法来恢复所需的溶剂化结构,并有望将其应用扩展到极低的温度。基于这些考虑,本工作将丁酸乙酯(EB)确定为要氟化的溶剂,因为其熔点低(图1b)。开发了一系列含有EB类似物作为共溶剂的高性能LIB电解质,包括具有不同氟化程度或氟位点的4,4,4-三氟丁酸乙酯(ETFB)、七氟丁酸乙酯和2,2,2-三氟丁酸乙基酯(TFEB)(图1c)。

【图2】a 静电势(ESP)图显示了本工作中考虑的溶剂分子电荷分布。b Li+与不同溶剂的结合能。c基础电解质和含有EB、ETFB、EHFB和TFEB的电解质电导率。d上述电解质的DSC曲线。e不同电解质在−110 °C下储存后的图像,持续30 min。f溶剂化结构与电解质物理性质的关系。
氟化EB衍生物在羰基O原子周围的电子密度低于EC或EB(图2a),导致与Li+的结合较弱(图2b)。此外,氟化程度影响助溶剂的电负性和Li+-溶剂配位强度(图2b)。例如,EHFB-Li+在含氟共溶剂中具有最低的结合能(~1.72 eV),并且在混合溶剂中EHFB系统的EC-Li+具有最低的结合能(0.71 eV)。这些结果表明,氟化不仅降低了丁酸乙酯本身的配位,而且削弱了周围EC和Li+之间的相互作用,后者可以用“偶极-偶极效应”来解释。具体地说,氟的强电子亲和力导致电荷分布从C=O端偏移到-CF3端,这改变了氟化丁酸乙酯和周围EC分子之间的偶极-偶极相互作用,最终导致EC对Li+的弱溶剂化。
弱溶剂化结构显著改善了电解质的物理性质(图2f),如离子电导率和流动性。这是因为(i)一些EC分子从溶剂化结构中释放,以及(ii)更多低熔点的DEC和共溶剂参与溶剂化结构,导致较低的熔点(图2d)和较低的体相电解质粘度(图2c插图)。具体而言,估计冰点增加的顺序为EHFB(−135 °C),TFEB(−132 °C)和ETFB(−130 °C)(图2d),遵循与EC-Li+结合能相同的趋势。EHFB电解质即使在−110 °C下储存30分钟依然能保持液态(图2e)。基于粘度效应和介电效应(图2f),高离子电导率取决于的高介电常数和低粘度,因此具有高介电常数和低粘度的EHFB电解质在−90 °C下实现了约1.46 mS·cm−1的高离子电导率 (图2c)。相反,基础电解质在约−50 °C时冻结(图2d),当温度降至−90 °C时,显示出较差的离子导电性(0.001 mS·cm−1)(图2c)。

【图3】基于分子动力学(MD)模拟计算出−70°C下基础电解质(a)和EHFB电解质(b)的径向分布函数(g(r))和配位数(n(r)。c 在EHFB电解质中不同温度下EC和DEC的FTIR光谱C=O峰区域。d用Voigt函数拟合−70 °C下EHFB电解质的FTIR光谱。基础电解质(e)和EHFB电解质(f)中EC的对称环变形和(C–O)键的拉伸振动拉曼光谱。g基于计算的RDF,五种电解质在25 °C时的配位数。h 25 °C和−70 °C下基础电解质和EHFB电解质中所有溶剂组分与Li+的配位数。i在不同温度下,基础电解质和EHFB电解质中的溶剂化EC与游离EC以及溶剂化EC与溶剂化DEC的比值。J当温度从25°C降低至−70 °C时,基础电解质和EHFB电解质中溶剂化结构变化示意图 。
通过分子动力学(MD)模拟将溶剂化结构解耦,并提供了在每种电解质情况下计算的Li+的径向分布函数(RDF)(g(r))和配位数(n(r))(图3a、b)。每种溶剂的第一溶剂化半径为~2.65 Å,PF6−占据第二个溶剂化壳层(~4.2 Å)形成分离的离子对(SIP-PF6−)。在基础电解质中,EC是第一Li+溶剂化壳中的主溶剂,室温下的配位数为1.31,然后是线性碳酸盐DEC(1.13)和EMC(0.94)(图3g)。然而,随着上述共溶剂的加入,Li+溶剂化结构发生了显著变化。具体而言,EHFB电解质中的EC配位数急剧下降至0.61(图3b和图3g),而DEC配位数上升至1.46(图3b和图3g)。在EB、ETFB和TFEB系统中观察到类似的趋势,表明从EC主导的溶剂化结构转变为DEC主导的溶剂化结构。这些转变与氟化相关,并且共溶剂的氟化度越高,DEC溶剂与Li+的配位就越强,EC与Li+之间的相互作用就越弱。这些影响在较低的温度下更为明显。例如,EHFB电解质中的DEC配位数从25°C时的1.46增加至−70 °C时的1.71,而EC配位数从0.61下降到0.33(图3h),表明在LT的第一个溶剂化壳中,更多的EC被DEC取代。相反,在基础电解质中,随着温度的降低,EC与Li+的配位数增加(从25°C时的1.34增加到-70°C时的1.76),而DEC配位数减少(从25°C时的1.13减少到-70°C时的0.74)。考虑到配位每个Li+的溶剂分子总数恒定,EC配位的减少不可避免地导致DEC配位的增加。显然,减少EC配位的优势不会被DEC配位的增加所掩盖,这归因于DEC相对于EC固有的低极性性质。一方面,由于EC是一种高极性溶剂,因此EC配位数的减少无疑会导致溶剂化的减弱。另一方面,DEC的配位数的增加也导致溶剂化的减弱,因为其极性较低。因此,这两个因素协同促进了整体弱化溶剂化结构的形成。这突出了EHFB共溶剂在LT应用中的优点。除了氟化共溶剂对主要溶剂溶剂化的影响外,它们还影响Li+溶剂化,因为EB、ETFB、TFEB和EHFB的配位数随着氟化的增加从1.3、0.85、0.74降至0.06(图3g)。这与计算的Li+与溶剂结合能十分一致(图2b)。此外,EHFB的配位数从25 °C时的0.06急剧增加到−70 °C时的0.56(图3h)。这表明EHFB在LT下对Li+的溶剂化更具活性。低ε溶剂与共溶剂一起主导了溶剂化结构,导致Li+与内溶剂化鞘中所有溶剂之间的相互作用减弱(图3j)。这些特征促进了Li+的脱溶剂化,从而增强了LT下的电化学动力学。
温度相关的FTIR和拉曼光谱分析进一步证实了共溶剂与主溶剂一起对Li+溶剂化的贡献(图3c-f)。游离和溶剂化C=O的IR峰(在1660-1870 cm−1的区域内)用Voigt函数拟合(图3d)。溶剂化EC与游离EC之比(R1)和溶剂化EC与溶剂化DEC之比(R2)(等式1和等式2)用于量化溶剂化EC的相对丰度以及EC和DEC之间的配位竞争。
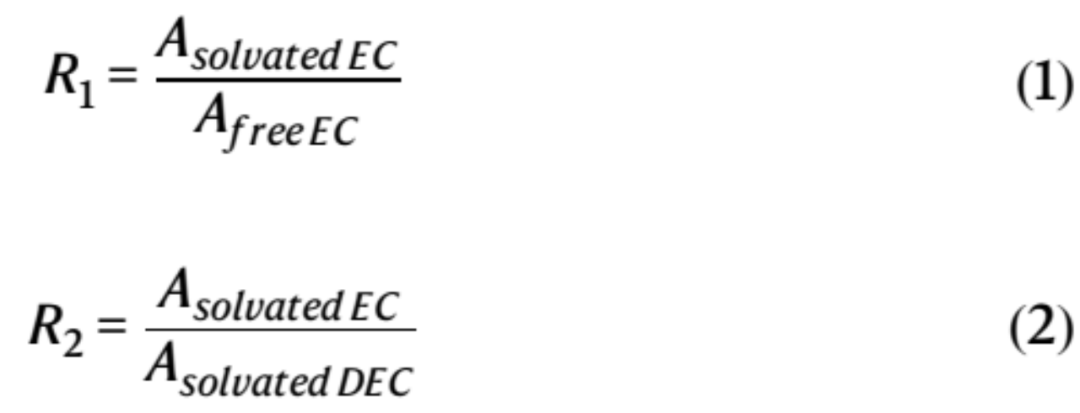
其中Asolvated EC、Afree EC和Asolvate DEC是溶剂化EC、游离EC和溶剂化DEC的C=O基团对应的振动峰积分面积强度(图3d)。定量分析显示,随着温度从25℃下降至−70 °C,R1和R2值都有所下降(图3i)。例如,EHFB电解质的R1值从1.34(25 °C)下降至1.09(−70 °C),同时R2从1.53(25 °C)下降至0.66(−70 °C)(图3i)。这表明,共溶剂促进了DEC和Li+之间的配位,同时EC和Li+间的相互作用减弱,尤其是在LT。注意,与ETFB和TFEB相比,EHFB电解质的R2值下降得更快(图3i),表明其在LT下对DEC主导的溶剂化结构有更大的选择性。相反,对于基础电解质,R1和R2都随着温度从25 °C降低至−70 °C而增加,证实了在宽温范围内EC主导的溶剂化作用(图3i)。此外,这种变化伴随着拉曼光谱中两个溶剂化EC峰的显著蓝移(从741到749 cm−1,从904到911 cm−1)(图3e),进一步表明在LT基础电解质中,Li+和EC之间的配位相互作用更强。这降低了其在寒冷条件下的实用性。这些特征,包括降低的R1和R2值(图3i)和抑制的C-O峰蓝移(图3f),证明共溶剂能够减弱EC的配位强度,从而设计了以DEC为主溶剂,共溶剂参与的溶剂化结构,最终形成了弱溶剂化结构(图3j)。
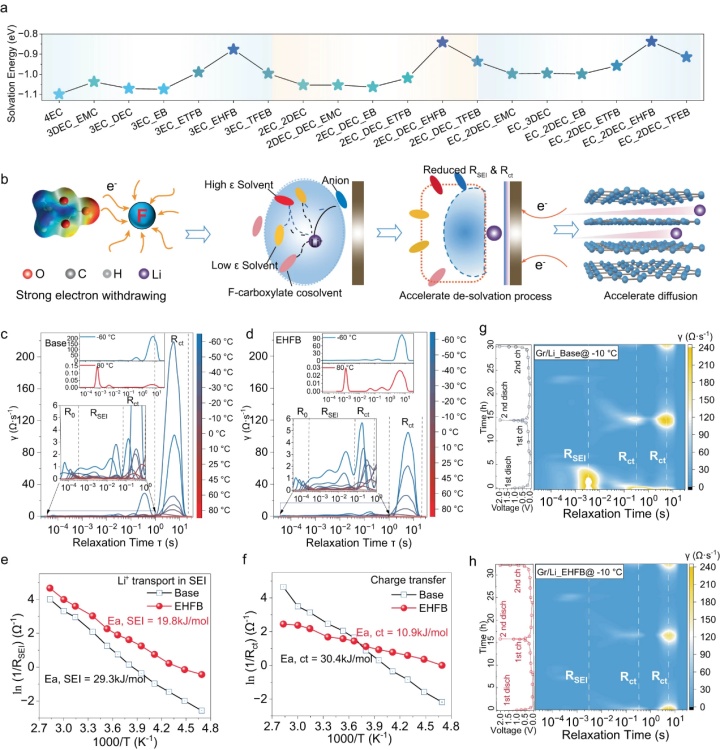
【图4】a不同电解质中的Li+溶剂化/脱溶剂化能。b氟化共溶剂在削弱Li+和溶剂分子之间的相互作用、加速脱溶剂化和促进Li+扩散方面的关键作用。根据采用基础电解质(c)和EHFB电解质(d)的LCO/Gr软包电池EIS数据得出的弛豫时间分布(DRT)的温度依赖性图。通过Arrhenius拟合Rct(e)和RSEI(f)得到相应的活化能。−10 °C下,采用基础电解质(g)和EHFB电解质(h)的石墨/Li半电池两个循环的原位DRT数据。
DFT计算表明,氟化共溶剂显著降低了EC/DEC电解质的Li+脱溶剂化能(图4a),并且这一趋势高度依赖于氟化。共溶剂的氟化程度越高,脱溶剂化能越小(从4EC的1.1 eV至EC-2DEC-EB的1.0 eV,EC-2DEC-ETFB的0.93 eV和EC-2DEC-EHFB的0.83 eV)。这是由于氟取代基的强吸电子效应,增强了Li+和低ε溶剂之间的配位,并形成了DEC主导的、总体减弱的溶剂化结构,从而促进了脱溶剂化过程(图4b)。
通过对弛豫时间(DRT)分布分析进一步验证了氟化共溶剂对Li+脱溶剂化的影响(图4c,d)。该分析通过连续分布函数中的局部最大值对不同的电化学过程进行了分类。Rct和RSEI都表现出强烈的温度依赖性。低温时Rct占主导地位,高温时RSEI占主导地位。具体而言,EHFB电解质在−60°C时具有1.0 Ω的最低Rct(图4d),然后是TFEB(3.2Ω)和ETFB(4.7Ω)。这些值远小于基础电解质(8.8Ω)(图4c),表明助溶剂在促进电荷转移中的重要作用。电荷转移的活化能较低也证实了这一点(图4f),并且具有EHFB电解质的LCO/Gr软包电池活化能估计为10.9 kJ·mol−1,约为基础电解质的三分之一(30.4 kJ·mol−1)。活化能按EHFB<TFEB< ETFB的顺序增加,与脱溶剂化能的趋势非常一致(图4a)。采用基础电解质和EHFB电解质的Gr/Li半电池在−10°C下两次电化学循环的DRT图谱(图4g,h)表明,EHFB系统表现出比基础电解质小得多的Rct,证实了共溶剂的引入促进了电荷转移,特别是在低温运行的石墨负极中。此外,与基础电解质(29.3 kJ·mol−1)(图4e)相比,具有EHFB的全电池中Li+通过SEI的活化能低得多(19.8 kJ·mol−1)(图4f),因此RSEI显著降低(图4g,h)。此外,与基础电解质不同(图4g),EHFB系统中的RSEI略有变化,其值要小得多(图4h),这表明源自EHFB的SEI具有高导电性,并且比基础电解质中的SEI更坚固。
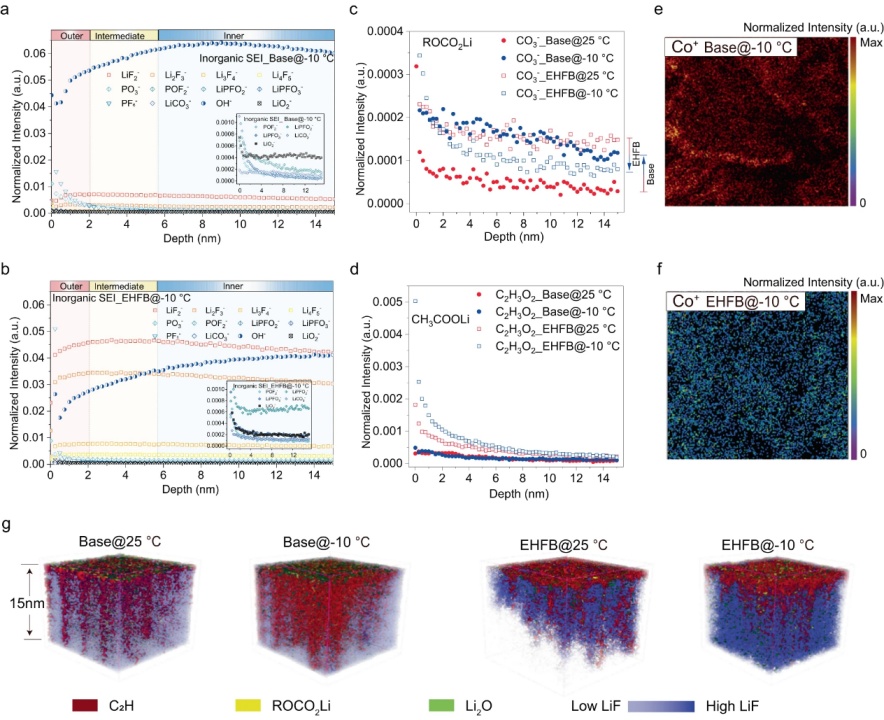
【图5】在基础电解质(a)和EHFB电解质(b)中,−10°C下长循环后无机SEI中官能团的深度分布。25 °C和−10 °C时基础电解质和EHFB电解质形成的SEI中ROCO2Li组分(c)和CH3COOLi组分的深度分布 。采用基础电解质(e)和EHFB电解质(f)的石墨负极上Co+的空间分布。g在25°C和-10°C下,采用基础电解质和EHFB电解质的电池化成后石墨负极SEI最上面15nm的三维重建图。
用飞行时间二次离子质谱法(TOF-SIMS)表征了循环石墨电极上SEI层的成分/结构演变。深度剖面TOF-SIMS数据显示,有机部分(CH2−、CO3−、C2H3O−和C2H3O2−)主要集中在SEI的外表面(图5c、d),而无机LiF物种(LiF2−、Li2F3−、Li3F4−、Li4F5−)和OH−在SEI内侧更普遍(图5a、b)。离子碎片潜在来源包括三种主要有机成分:ROCO2Li(CO3−碎片)、CH3COLi(C2H3O−)和CH3COOLi(C2H3O2−),它们分别通过EC、DEC和羧酸盐的电化学还原产生。在基础电解质中,ROCO2Li信号随着温度从25 °C降低至−10 °C而增强(图5c),而CH3COLi信号减少,并且几乎没有检测到CH3COOLi信号(图5d)。这表明在基础电解质中EC是LT下进行还原的主要溶剂,而不是DEC。相反,−10 °C下,当使用EHFB电解质时,ROCO2Li信号降低,CH3COLi信号增加(图5c),表明在EHFB中,LT增强了DEC还原并抑制了EC还原。在基础电解质中,EC对Li+的溶剂化在LT时得到促进,但在EHFB情况下得到缓解。此外,检测到的CH3COOLi信号表明,EHFB共溶剂参与LT时石墨表面的成膜(图5d)。此外,在源自EHFB电解质的界面中检测到更多的LiF物种(图5a,b),并且它们的量随着温度的降低而增加。SEI性质的差异反映在不同温度下有机SEI组分(C2H,ROCO2Li)和无机物种(LiF,Li2O)的3D空间分布覆盖图中(图5g)。具体而言,在EHFB中,仅在SEI的最外层中发现少量有机组分(即C2H),并且LT下更多的LiF存在于SEI内层中。这可能是由于更多的EHFB参与溶剂化并有助于LT时SEI的形成。相反,基础电解质衍生的SEI中LiF少得多(图5a,图5d)。此外,在EHFB电解质中,RT和LT条件下Co+的溶解和串扰被有效抑制(图5e、f)。这可能是由于氟化电解质的抗氧化性,用它可以很好地保护正极结构在约4.5 V的高电压下不受损坏。这突出了氟化共溶剂在高压应用中的显著优势和SEI的优异性能。相反,在基础电解质中,大量的Co+被溶解,然后串扰到石墨负极,这对SEI具有高度破坏性(图5e、f)。
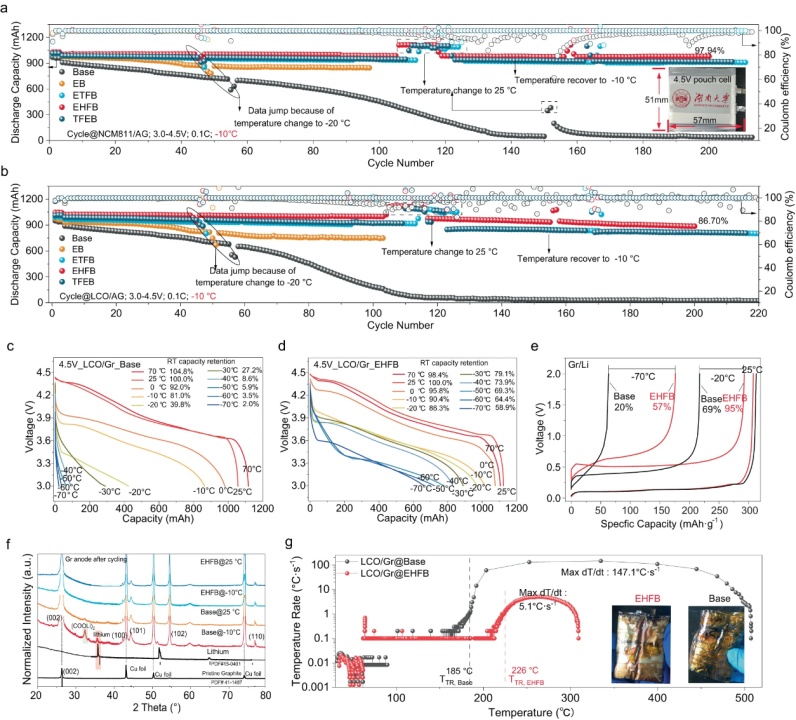
【图6】NCM811/Gr(a)和LCO/Gr(b)电池在−10°C下不同电解质中的循环行为。具有基础电解质(c)和EHFB电解质(d)的LCO/Gr软包电池RT充电-LT放电的电压曲线。e具有不同电解质的Gr/Li电池在不同温度下的充电电压曲线。f在−10°C和25 °C下长循环后石墨的XRD图谱。g完全充电LCO/Gr电池的温升速率随温度的变化。插图显示了测试后的照片。
将EHFB电解质与商业1Ah NCM811/Gr和LCO/Gr软包电池匹配获得了优异的低温电化学性能。在−10 °C下200次循环后,NCM811/Gr的容量保持率为97.94%,LCO/Gr的容量保持率为86.70%(图6a,b)。利用RT充电-LT放电方案,使用EHFB电解质的LCO/Gr软包电池在−40 °C时保持830 mAh的高容量 ,相当于其RT容量的73.9%。即使在−70°C时,这些电池仍保留了约60%的RT容量(图6d)。相反,使用基础电解质的LCO/Gr和NCM811/Gr电池表现出较差的性能,在−10 °C时仅保留约80%的RT容量 ,在−40 °C时完全失效 (图6c)。电池的低温性能受到石墨负极的限制,这是由锂枝晶引起的。事实证明,当在−10 °C下循环时,石墨负极基电池表现出显著的容量衰减 (图6a,b)。因此,对Gr/Li电池在低温下的性能进行了评估。采用EHFB电解液的Gr/Li电池在-70°C和-20°C下充电,室温放电后分别保持了57%和95%的容量。这些值远高于基础电解质的值(分别为20%和69%的RT容量,图6e)。XRD分析显示,在−10°C循环的石墨负极中,在35.8°处观察到明显的锂峰(图6f),而在EHFB情况下不存在。这些发现都证实了改性电池在低温下能有效抑制锂枝晶的形成。使用加速量热法(ARC)评估安全性能(图6g)。结果表明,与基础电解质相比,使用EHFB电解质的LCO/Gr软包电池安全性显著提高。EHFB电解质导致最大dT/dt显著降低,仅为5.1 °C s−1和高得多的热失控温度(TTR,226 °C),能够有效避免热失控事件。此外,最高温度降至309 °C,表明热失控过程中释放的总能量大幅减少。
总结和展望
本工作展示了一种使用传统EC基电解质的溶剂化设计策略,以提高LIBs低温性能。通过引入氟化共溶剂,削弱了EC与Li+的强配位,并将EC主导的溶剂化结构转变为DEC主导的溶剂化结构,特别是在低温下。这种设计促进了Li+的脱溶剂化,同时保持了EC的高介电性能。此外,氟化共溶剂也有助于形成富氟SEI,增强了石墨负极在寒冷条件下的稳定性。因此,在低温性能方面取得了显著的改进,例如更宽的流动性范围(将液体保持在−110 °C),电导率更高(在−90 °C时为1.46 mS·cm−1 ),更容易的脱溶剂化过程,并抑制了锂枝晶的生长。这使得1 Ah 4.5 V石墨基软包电池在−10 °C下稳定循环200多次,当在−60 °C下充放电一圈时,只有2%的容量损失并保持334 mAh的容量。此外,电池在−70°C时放电 ,仍有60%的室温容量,并能够在约−100 °C的极低温度下为设备供电 。这项工作为开发适用于极端环境的锂离子电池提供了一种独特的方法。
参考文献
Yuqing Chen, Qiu He, Yun Zhao, Wang Zhou, Peitao Xiao, Peng Gao, Naser Tavajohi, Jian Tu, Baohua Li, Xiangming He, Lidan Xing, Xiulin Fan & Jilei Liu*. Breaking solvation dominance of ethylene carbonate via molecular charge engineering enables lower temperature battery, Nature Communications.
DOI:10.1038/s41467-023-43163-9
https://doi.org/10.1038/s41467-023-43163-9
注:图片非商业用途,存在侵权告知删除!
本文地址:https://libattery.net/news/details1886.html
好文章,需要你的鼓励
邮箱:libatterychina@163.com
北京:北京市海淀区上地三街9号金隅嘉华大厦C座904
010-62980511
山东:山东省临沂市鲁商中心A12-1503-1
0539-8601323
锂电中国(libattery.net)版权所有
Copyright By 北京贝特互创科技有限公司
京ICP备11002324号-1
京公安网备11010802035676号

 手机锂电网
手机锂电网





我有话说: